Crystallography Phase Determination
Creative Biostructure offers high-throughput UniCrys™ X-ray crystallography services by using state-of-the-art facilities, and has developed a crystallography pipeline that includes all the stages from protein expression to structure determination. Our experienced scientists can teamwork with customers and accelerate the research progress.
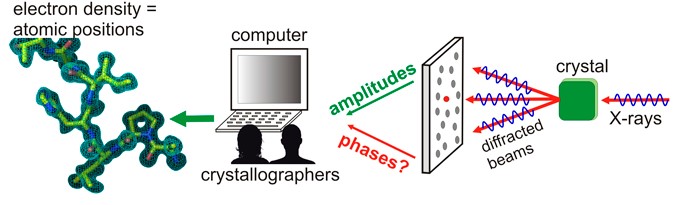 Figure 1. Phase determination from diffraction data
Figure 1. Phase determination from diffraction data
After successful crystallization and the diffraction pattern is obtained with our protein crystallization services, the next challenge is to get an electron density map from which detailed information about the structure can be deduced. This is only possible if the ‘phase problem’ can be solved. Each reflection on the diffraction pattern corresponds to a wave consisting of an amplitude and a phase. The amplitude of diffraction spots can be simply measured by taking the square root of the intensity that X-ray detector records. However, the information on the phases, which contain critical information for determining the electron density distribution in the unit cell, is lost during data collection. The structure of small molecules may be solved in the absence of phase information, but this is not true for macromolecules such as proteins.
Our scientists at Creative Biostructure are highly experienced with phase determination. We use a variety of methods to find initial phases that can subsequently be refined (see Table 1).
Table 1. Protein crystallization services at Creative Biostructure
| Service | Feature |
Molecular Replacement (MR) | MR is used when the protein under study has a homolog (sequence identity > 40%) whose structure is known. In this case, initial phases can be approximated from the known structure by its rotation and translation in the unit cell. Once the correct orientation and translation are identified, the model is used to calculate phases for all the structure factors. Electron density maps may then be calculated using phases from the model structure and weighted magnitudes from the unknown one. |
Multiple Isomorphous Replacement (MIR) | MIR is a way of determining phases by soaking the crystal in a heavy atom solution and then comparing the diffraction pattern of this derivative crystal to that of the native crystal. The heavy atoms scatter more strongly than other atoms in the structure and therefore contribute enormously to the diffraction, which makes it easy to locate them and calculate their scattering both in magnitude and in phase. |
Single- or Multi-wavelength Anomalous Dispersion (SAD or MAD) | SAD or MAD involves the use of synchrotron radiation to excite the crystal at a single or multiple specific wavelengths. Most commonly, all methionine residues are replaced by selenomethionines in the protein. Selenomethionine contains a selenium instead of a sulfur atom, which makes it a moderately effective anomalous scatterer. Phase information may be recovered by the analysis of phase shift. This method requires only a single crystal and reduces potential radiation damage to the molecule. |
Depending on the quality of the phasing experiment, there can be rather large errors in the phases and thus in the electron density maps. Our scientists will apply their knowledge and expertise, and use a variety of techniques to refine the phases in an iterative manner.
The structural biology team at Creative Biostructure carries out high-resolution structure determination by using both in-house X-ray facility (for trial and optimization) and synchrotron light source (for data collection). High-resolution X-ray data, high-quality density maps, protein structures and statistics will also be made available for the purpose of publication and subsequent studies.
Please feel free to contact us for a detailed quote.
Ordering Process

References
- Wolf E (2009). “Solution of the phase problem in the theory of structure determination of crystals from X-ray diffraction experiments”. Phys Rev Lett 103(7):075501.
- Kleywegt GJ (2000). “Validation of protein crystal structures”. Acta Crystallogr D Biol Crystallogr 56(Pt 3):249–265.


