Molecular Dynamics Simulation for the Antiviral Drug Discovery of Coronavirus
Molecular dynamics (MD) simulation is an in-silico simulation technique applied to study the physical movements of atoms and molecules. It enables one to use the laws of physics to predict the time-dependent evolution of interacting particle systems. In this approach, atoms and molecules are allowed to interact within a given period of time, thus giving a view of the dynamic evolution of the system. The static models generated by X-ray crystallography, NMR spectroscopy, and homology modeling provide a basic three-dimensional (3D) structural model for biomolecular molecules, while MD simulations can offer atomic-level information about the conformational changes and binding thermodynamics of biomolecular molecules under predetermined physiological conditions. Creative Biostructure utilizes MD simulation methods to help customers predict changes in protein systems involved in coronavirus-related studies over time or under predefined physiological conditions, thereby accelerating the research of new antiviral drugs.
Brief Introduction to Coronavirus and SARS-CoV-2 Infection
Novel coronavirus infection (COVID-19) has spread to all parts of the world since the first case was reported in Wuhan, China in 2019. And the world health organization (WHO) announced that the outbreak to be a global pandemic. The genome of the coronavirus is a single-stranded RNA. After infecting the host cell, polyproteins are translated through the RNA genome. As the polyprotein is cleaved, the fragments will function as structural proteins and enzymes needed for virus propagation. The main protease (Mpro, also termed as 3-chymotrypsin-like protease, 3CLpro) that catalyzes the cleavage of the polyprotein is one of the target proteins. Scientists are looking for inhibitors that bind to the protease and have previously analyzed the X-ray crystallographic structure of small molecules binding to SARS-CoV-2 Mpro. Recently, it was reported that scientists used a supercomputer to simulate the structural dynamics of SARS-CoV-2 Mpro in 10 microseconds. This data can be used to calculate the Mpro structural dynamics before drug molecule binding and predict the binding strength of candidate molecules to the protease.
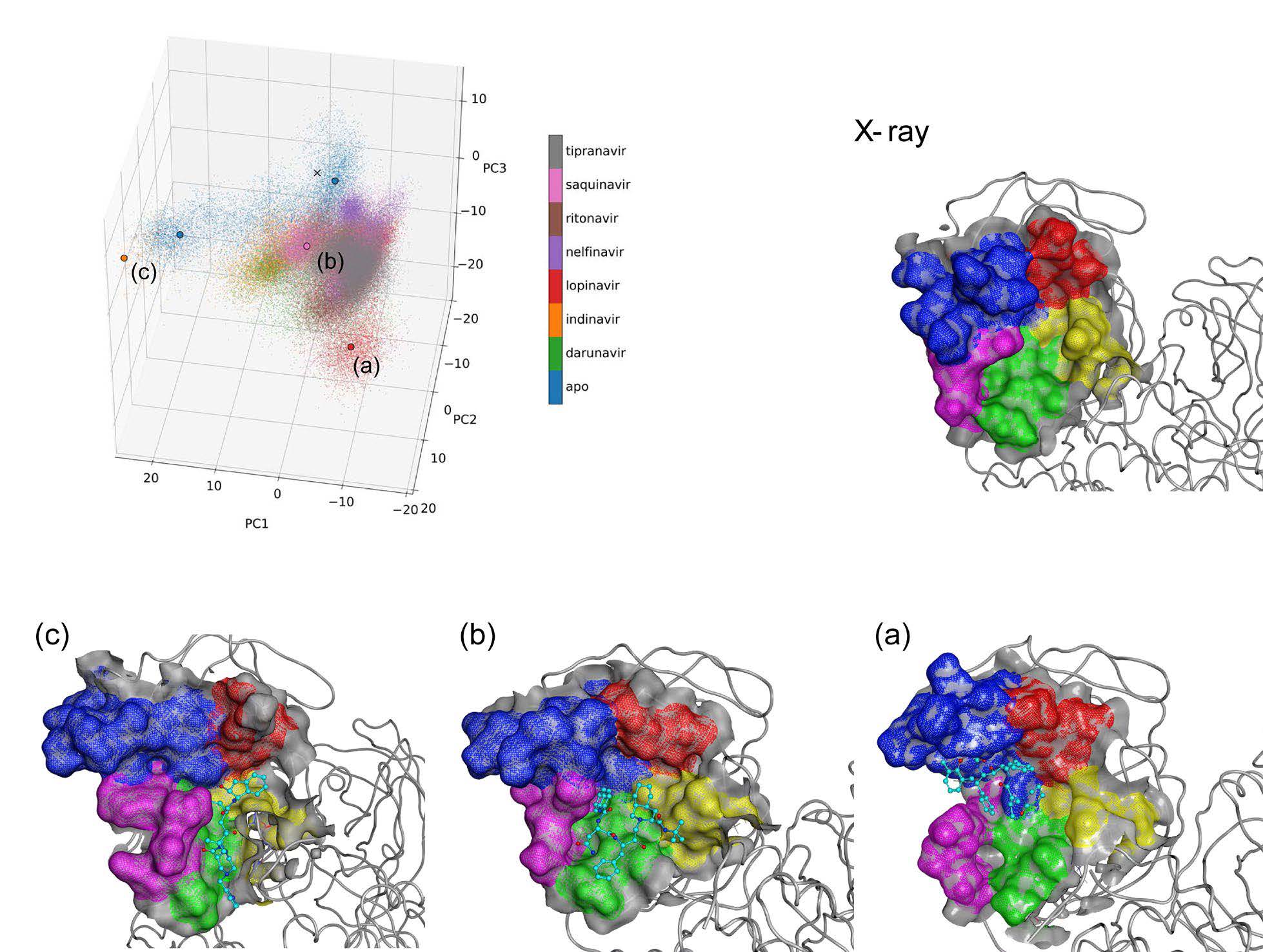 Figure 1. Conformational diversity of the Mpro active site. (KOMATSU, Teruhisa S.; et al. 2020)
Figure 1. Conformational diversity of the Mpro active site. (KOMATSU, Teruhisa S.; et al. 2020)
Molecular Dynamics Simulation Services for the Antiviral Drug Discovery of Coronavirus
MD simulation is particularly useful in computer-aided drug discovery (CADD) for the identification of hidden or allosteric binding sites, enhancement of traditional virtual screening methods, and direct prediction of the binding energy of small molecules. Creative Biostructure exploits state-of-the-art software tools to provide customers with MD simulation services for studying the molecular dynamics of protein systems involved in the process of coronavirus infection.
The general procedure of our MD simulation service consists of the following steps: 1) Select a model system, in which the missing segments are fixed and the protonation states determined; 2) By solving Newton’s equation of motion, the energy of the system is minimized and equilibrated until the properties of the system no longer change with time; 3) The production run will be executed in the appropriate time frame to output trajectories after equilibration, which will then be analyzed to obtain the desired characteristics.
With more than 10 years of experience in structure-based drug design (SBDD), we are willing to provide our customers with the optimal MD simulation services. If your coronavirus-related research project has such research needs, please feel free to contact us, and our customer service representative is available 24 hours a day from Monday to Sunday.
Contact us to discuss your project!
References
- Zhang L.; et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020.
- KOMATSU, Teruhisa S.; et al. Drug binding dynamics of the dimeric SARS-CoV-2 main protease, determined by molecular dynamics simulation. ChemRxiv. Preprint. 2020.
