Quick Overview – Linear vs Conformational Epitope at a Glance
| Characteristic | Linear Epitopes | Conformational Epitopes |
|---|---|---|
| Structure | Consist of continuous amino acid sequences in the protein's primary structure | Formed by amino acids that are distant in sequence but brought together by protein folding |
| Sequence Nature | Sequential, adjacent residues (typically 5-15 amino acids) | Discontinuous residues from different parts of the protein |
| Stability | Highly stable under denaturing conditions (heat, extreme pH) | Easily destroyed by conditions that disrupt protein folding |
| Preservation Method | Can be represented by synthetic peptides | Require preservation of protein's tertiary structure |
| Recognition After Denaturation | Remain recognizable when protein is denatured | Lost when protein structure is disrupted |
| Prevalence in Nature | Minority of naturally occurring epitopes (~10%) | Majority of B-cell epitopes in natural immune responses (~90%) |
| Immunogenicity | Generally lower in native state, but can be enhanced artificially | Higher in natural immune responses |
| Binding Specificity | Generally lower specificity | Higher specificity and affinity |
| Mapping Complexity | Relatively straightforward to map | More challenging to characterize |
| Primary Mapping Techniques | Peptide arrays, phage display, bacterial display | X-ray crystallography, cryo-EM, HDX-MS, site-directed mutagenesis |
| Applications | Immunoblotting, some diagnostics, peptide vaccines | Therapeutic antibody development, functional inhibition |
Introduction to Epitopes
Epitopes are specific portions of antigens that are recognized by antibodies and play a critical role in immune responses. These molecular structures serve as binding sites for antibodies and B cell receptors, making them essential for the development of vaccines, diagnostics and therapeutics.
Epitope mapping identifies the precise regions of antigens that interact with antibodies, providing valuable insight into immune recognition. Understanding the differences between linear and conformational epitopes is fundamental to selecting appropriate mapping techniques and applications in biomedical research.
 Figure 1. Schematic diagram of linear epitopes and conformational epitopes. (Creative Biostructure)
Figure 1. Schematic diagram of linear epitopes and conformational epitopes. (Creative Biostructure)
What Is a Linear Epitope
Linear epitopes (also called continuous epitopes) consist of sequential amino acids within a protein's primary structure. Typically spanning 5-15 consecutive amino acid residues, these epitopes form a continuous segment along the protein backbone, making them relatively straightforward to identify.
Advantages of Linear Epitopes
- Easier to identify through sequence-based techniques
- Often retain immunogenicity when isolated from the complete protein
- Suitable for peptide-based vaccine development
Limitations of Linear Epitopes
- Represent only a minority of naturally occurring epitopes
- May not mimic the three-dimensional structure in native proteins
Common Applications in Immunoblotting and Diagnostics
Monoclonal antibodies recognizing linear epitopes are preferred for immunoblotting because these techniques often involve denatured proteins where conformational epitopes are disrupted. Under denaturing conditions (such as SDS-PAGE), proteins lose their tertiary structure, but linear epitopes remain detectable. In diagnostics, linear epitopes serve as valuable biomarkers for various diseases, particularly in ELISA tests and other immunoassays using synthetic peptides.
What Is a Conformational Epitope
Conformational epitopes (discontinuous epitopes) are formed by amino acid residues that are distant in the protein's primary sequence but brought into proximity through protein folding in the tertiary structure. This three-dimensional arrangement creates a unique topographical surface recognized by antibodies.
Importance in Therapeutic Antibody Development
Conformational epitopes are crucial in therapeutic antibody development because:
- Over 90% of B-cell epitopes are conformational in nature
- Therapeutic antibodies often target proteins in their native, folded state
- These epitopes frequently include functional regions of proteins, such as binding pockets or enzymatic active sites
Challenges in Identification and Characterization
Despite their prevalence, conformational epitopes present significant challenges:
- They are lost when proteins are denatured
- Cannot be easily represented by short peptide fragments
- May undergo subtle changes depending on environmental conditions
- Require sophisticated techniques for analysis
Comparison of Linear vs Conformational Epitopes
Structural Differences
Linear epitopes consist of sequential amino acids, forming a continuous segment. Conformational epitopes comprise residues from multiple regions of the protein, creating a complex topographical interface for more specific antibody recognition.
Stability Under Different Conditions
Linear epitopes generally show greater stability under denaturing conditions (high temperature, extreme pH). Conformational epitopes are highly sensitive to conditions that alter protein folding, as denaturation destroys the three-dimensional arrangement of amino acids.
Immunogenicity Differences
Conformational epitopes typically display higher immunogenicity in natural immune responses, as they better represent native structures encountered by the immune system. Linear epitopes can become highly antigenic when properly presented, making them valuable for vaccine design.
Role in Antibody-Antigen Binding
Antibodies recognizing linear epitopes engage with a sequential stretch of amino acids through basic interactions. Antibodies targeting conformational epitopes interact with a three-dimensional surface including amino acids from different parts of the protein sequence, resulting in higher binding specificity and affinity.
Linear Epitope Mapping Techniques
Peptide Arrays and Microarrays
Peptide arrays involve synthesizing overlapping peptide fragments spanning the entire target protein sequence. These peptides, typically 8-20 amino acids in length with overlaps of 5-10 residues, are immobilized on solid supports such as glass slides or membranes. When antibodies are applied, they bind to peptides containing their epitope sequences, which can be detected through visualization methods including fluorescent labels or enzymatic reactions.
Advantages and Applications of Peptide Arrays
A key strength of peptide arrays is their ability to analyze complex antibody mixtures, including polyclonal sera from samples. This makes them valuable for infectious disease research, autoimmunity studies, and vaccine development. The high-throughput nature allows systematic screening of entire proteins or even proteomes with minimal sample consumption.
Limitations of Peptide Arrays
Despite their utility, synthetic peptides may not replicate the conformations found in native proteins, leading to both false positives and negatives. Additionally, peptide arrays exclusively detect linear epitopes, missing the conformational epitopes that represent the majority of antibody-antigen interactions in natural settings.
Phage Display
Phage display technology expresses peptide libraries on bacteriophage surfaces. Libraries are screened against antibodies of interest, and binding peptides are isolated through biopanning. This technique can identify both linear epitopes and mimotopes that structurally mimic conformational epitopes.
System and Library Construction
Phage display utilizes bacteriophages genetically engineered to express peptide sequences on their surface proteins. Most commonly, filamentous phages like M13 display peptides fused to their pIII or pVIII coat proteins. Library construction involves inserting random DNA sequences into the phage genome, creating collections with diversities reaching billions of unique peptide variants.
Biopanning Process
The epitope mapping process, called biopanning, involves several cycles of selection and amplification. Phages are incubated with immobilized antibodies, non-binding phages are washed away, and bound phages are eluted, amplified in bacteria, and subjected to additional rounds of selection. This progressively concentrates phages displaying peptides with the highest antibody affinity.
Unique Features of Phage Display
Phage display offers several advantages, including identification of mimotopes (peptides structurally mimicking conformational epitopes), ability to work with low-affinity interactions, self-amplification of positive hits, and simultaneous screening of billions of candidates. However, it requires specialized expertise and can show selection biases for certain sequences.
Bacterial Display
Similar to phage display, bacterial display expresses peptides on bacterial surface proteins. Selection typically employs fluorescence-activated cell sorting (FACS) to isolate bacteria displaying antibody-binding peptides. This approach allows expression of larger peptides and quantitative assessment of binding affinity through flow cytometry.
Bacterial display systems can express peptides up to 100+ amino acids in length and support complex antigen presentation. The use of flow cytometry for screening enables researchers to establish quantitative affinity thresholds and isolate binders across a range of affinities. However, library sizes are typically smaller than phage display libraries.
Mass Spectrometry Approaches
Mass spectrometry (MS) offers several techniques for epitope mapping, including epitope excision (antibody-antigen complexes form before digestion), epitope extraction (antigen is digested first, then peptides are captured by antibodies), and hydrogen-deuterium exchange (measures protection from exchange when antibody binds).
A typical MS-based experiment involves complex formation between antibody and antigen, enzymatic digestion, isolation of protected peptides, and MS analysis to determine peptide identity. This approach offers high sensitivity, the ability to detect post-translational modifications, and works with native proteins under physiologically relevant conditions.
MS techniques can achieve single-amino-acid resolution but require sophisticated instrumentation, complex data analysis, and relatively high sample amounts compared to array-based methods.
In conclusion, each technique offers distinct advantages: peptide arrays provide high-throughput screening; phage and bacterial display offer diverse peptide libraries; mass spectrometry provides high sensitivity with native proteins. Combining multiple techniques often provides the most comprehensive linear epitope mapping.
Conformational Epitope Mapping Techniques
X-ray Crystallography
Methodology and Resolution
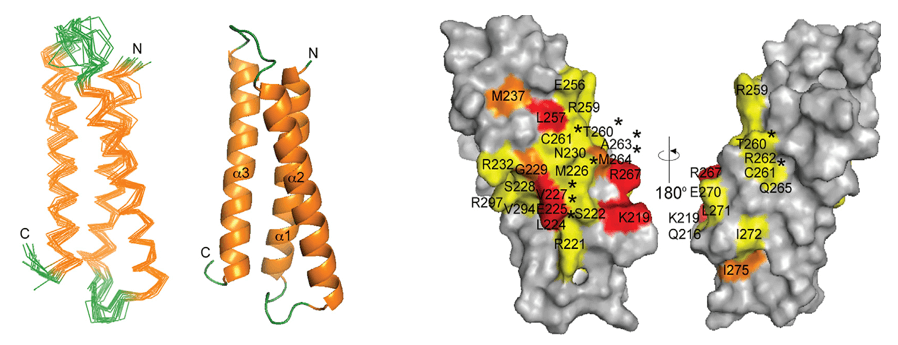
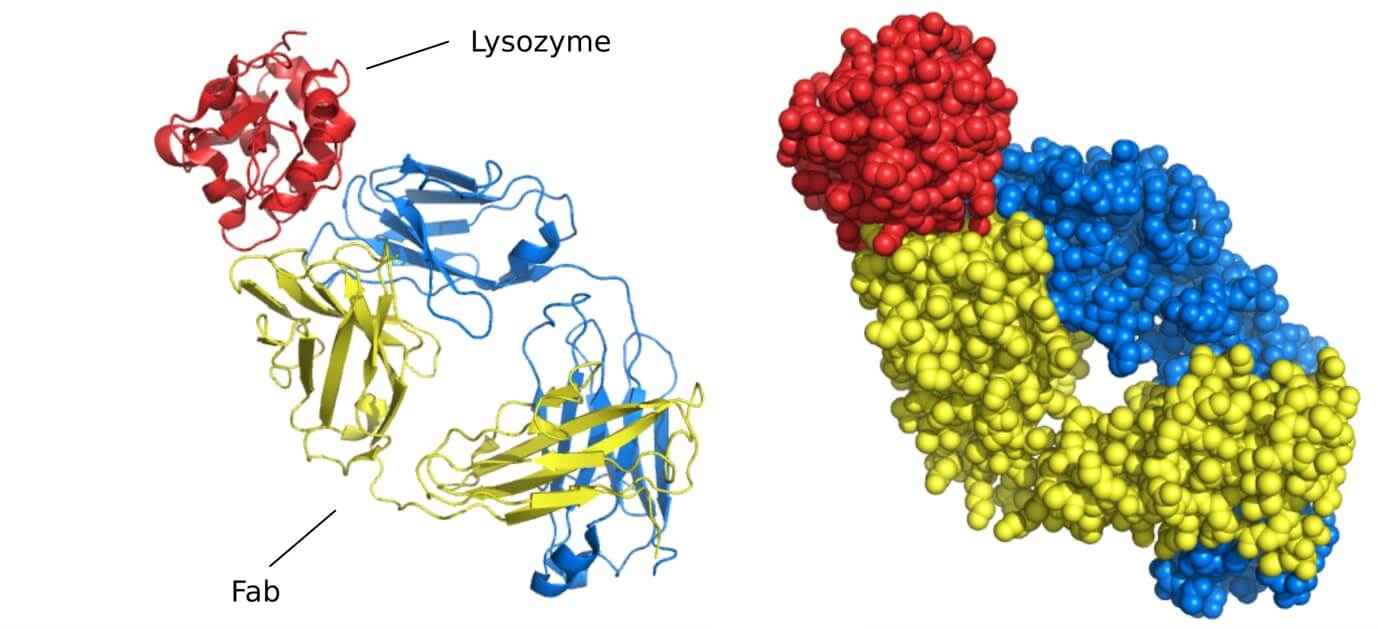
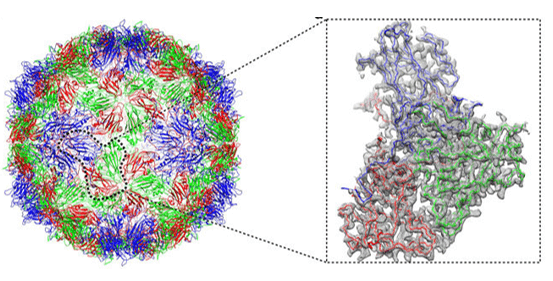
X-ray crystallography represents the gold standard for conformational epitope mapping, providing atomic-level resolution of antibody-antigen complexes. The technique involves crystallizing the antibody-antigen complex and analyzing the diffraction pattern created when X-rays pass through the crystal. Advanced synchrotron radiation sources enable resolutions below 2 Å, allowing researchers to visualize individual amino acid side chains and even water molecules at the binding interface.
Structural Insights
The exceptional detail revealed by crystallography has transformed our understanding of antibody-antigen interactions. Studies of crystallized complexes have demonstrated that conformational epitopes typically comprise 15-25 amino acids, with only 5-8 residues making energetically significant contributions to binding. These "hot spots" often cluster in protrusions or depressions on the antigen surface, forming complementary interfaces with the antibody paratope that involve an intricate network of hydrogen bonds, salt bridges, and van der Waals forces.
Practical Challenges
Despite its power, crystallography faces significant hurdles. Obtaining diffraction-quality crystals of antibody-antigen complexes is notoriously difficult, with success rates often below 30% even in specialized laboratories. The crystallization process may take months to years, requires milligram quantities of highly purified proteins, and sometimes introduces artifacts that do not reflect the dynamic nature of interactions in solution.
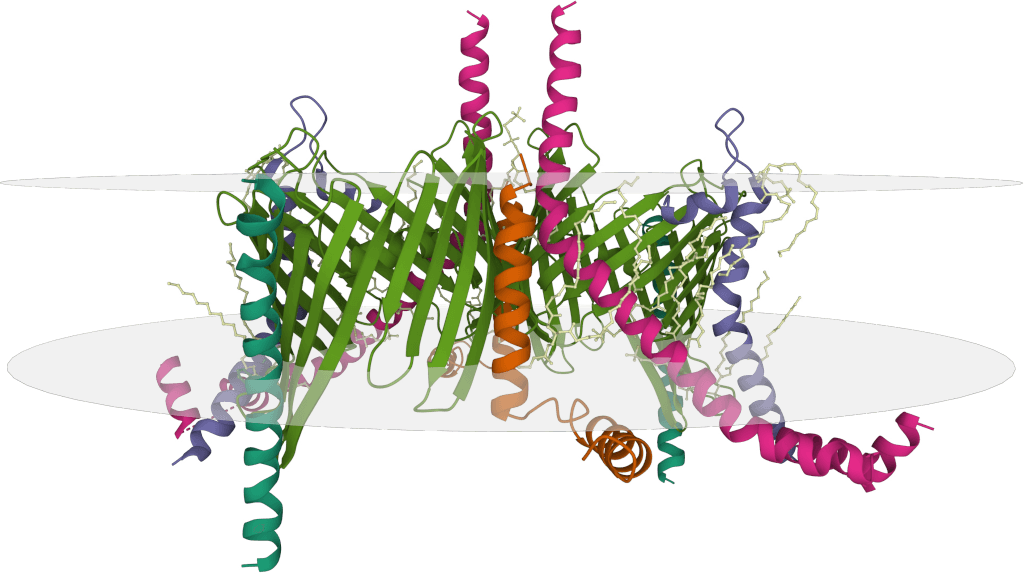
Cryo-electron Microscopy (Cryo-EM)
Technical Evolution
Cryo-electron microscopy has revolutionized conformational epitope mapping by enabling structural analysis without crystallization. The sample is flash-frozen in a thin layer of vitreous ice, preserving its native state, and imaged using an electron microscope. Direct electron detectors and improved image processing algorithms have dramatically enhanced resolution capabilities from >10 Å to near-atomic levels (2-4 Å).
Advantages for Complex Antigens
Cryo-EM excels at analyzing large macromolecular complexes and membrane proteins that resist crystallization. The technique can visualize multiple antibody binding states simultaneously, revealing conformational flexibility that may be critical for function. Recent studies have successfully mapped epitopes on challenging targets like viral glycoproteins, G-protein coupled receptors, and ion channels—structures that had previously eluded detailed analysis.
Methodological Considerations
While transformative, cryo-EM has limitations including the high cost of equipment, computational demands for image processing, and challenges in achieving high resolution for smaller proteins below ~150 kDa. Researchers often employ Fab fragments instead of full antibodies to reduce complex size and increase particle contrast, though this may occasionally alter binding characteristics.
Hydrogen-deuterium Exchange Mass Spectrometry (HDX-MS)
HDX-MS offers a solution-based approach to conformational epitope mapping by analyzing changes in protein dynamics upon antibody binding. The technique exploits the fact that hydrogen atoms in proteins exchange with deuterium at rates dependent on their solvent accessibility and involvement in hydrogen bonding. When an antibody binds to its epitope, it protects the binding site from deuterium exchange, creating a distinctive pattern detectable through mass spectrometry.
Experimental Process
A typical HDX-MS workflow involves exposing the antigen to deuterium-containing buffer both with and without antibody present, quenching the exchange reaction at various time points, digesting the protein into peptides, and analyzing these peptides by liquid chromatography-mass spectrometry. Regions showing differential deuterium uptake between bound and unbound states identify potential epitope components.
Practical Applications
HDX-MS has proven particularly valuable for epitope mapping when crystallography or cryo-EM are impractical. The technique requires less sample (typically 10-100 μg), accommodates structurally heterogeneous antigens, and provides information about binding-induced conformational changes throughout the protein, not just at the interface. However, spatial resolution is typically limited to peptide level (5-20 amino acids) rather than individual residues, and data interpretation requires careful consideration of protein dynamics.
Chemical Cross-linking Mass Spectrometry (XL-MS)
Chemical cross-linking mass spectrometry identifies conformational epitopes by covalently linking spatially proximate residues in the antibody-antigen complex. Various cross-linking reagents with different specificities and spacer arm lengths (typically 5-15 Å) are used to capture residues within defined distances. After cross-linking, the complex is enzymatically digested, and cross-linked peptides are identified through specialized mass spectrometry techniques.
Technological Implementation
XL-MS workflows incorporate photo-reactive cross-linkers that can be activated with UV light, zero-length cross-linkers that directly connect interacting residues, and MS-cleavable linkers that facilitate identification of cross-linked peptides. Advanced computational algorithms have significantly improved the detection and assignment of cross-links, enabling the mapping of complex interaction networks.
Strengths and Limitations
XL-MS offers several advantages, including the ability to capture transient interactions, compatibility with diverse protein types, and the capacity to work with complex sample mixtures. The technique provides distance constraints that can be integrated with computational modeling to develop structural models of the antibody-antigen complex. Limitations include the chemical specificity of cross-linkers (primarily targeting lysine and cysteine residues), potential for cross-linking-induced conformational changes, and the computational complexity of data analysis.
Site-directed Mutagenesis and Alanine Scanning
Site-directed mutagenesis provides a functional perspective on conformational epitopes by systematically altering potential epitope residues and assessing the impact on antibody binding. In alanine scanning, the most common implementation, individual amino acids are replaced with alanine (which eliminates side chain interactions while preserving backbone conformation), and binding affinity is measured through techniques like surface plasmon resonance or ELISA.
Interpretative Framework
Residues whose mutation significantly reduces antibody binding (typically causing >10-fold reduction in affinity) are identified as key epitope components. This approach directly links structural elements to their functional role in recognition, revealing energetic "hot spots" within the epitope. The analysis can be extended with other amino acid substitutions to probe specific interaction types or to engineer improved binding properties.
Implementation Considerations
While powerful, this approach requires expression and purification of multiple protein variants, making it labor-intensive for large-scale epitope mapping. The technique works best when guided by preliminary structural data or computational predictions to focus mutation efforts on likely epitope regions. Interpretation must also consider whether mutations disrupt protein folding rather than directly affecting antibody binding.
Computational Prediction Methods
Computational methods for conformational epitope prediction have advanced significantly, employing diverse algorithms from simple physicochemical property analysis to sophisticated machine learning models. These methods analyze protein features such as surface accessibility, hydrophilicity, sequence conservation, secondary structure propensity, and spatial clustering of residues to identify potential epitope regions.
Deep Learning Integration
Recent advances in deep learning have revolutionized epitope prediction by integrating multiple data types and learning complex patterns from known antibody-antigen complexes. Convolutional neural networks applied to 3D protein structures and graph neural networks capturing residue relationships have achieved prediction accuracies approaching 70-85% for well-characterized antigens, a dramatic improvement over earlier methods.
Practical Applications
Computational prediction serves as a valuable first step in epitope mapping campaigns, prioritizing regions for experimental validation and reducing the experimental search space. When integrated with even limited experimental data, prediction accuracy increases substantially. However, most algorithms still struggle with highly conformational epitopes, protein dynamics, and novel antigens with limited structural information, highlighting the continued importance of experimental validation.
The most successful conformational epitope mapping efforts typically integrate multiple complementary approaches. For example, computational prediction might identify candidate epitope regions that are then tested through targeted mutagenesis. HDX-MS or XL-MS can provide solution-phase validation of these interactions, with high-resolution structural techniques like cryo-EM or X-ray crystallography applied to the most promising candidates.
Advanced computational frameworks now enable the integration of diverse experimental data types, with each technique providing constraints that collectively define the epitope landscape. For instance, XL-MS distance constraints, HDX-MS protection factors, and mutagenesis binding data can be combined with molecular docking algorithms to generate high-confidence epitope models even when high-resolution structures are unavailable.
Select Service
Choosing the Right Epitope Mapping Approach: Linear vs. Conformational
| Consideration Factor | Linear Epitope Mapping | Conformational Epitope Mapping |
|---|---|---|
| Research Objective |
|
|
| Antigen Characteristics |
|
|
| Resource Requirements | Low to Moderate:
|
High:
|
| Sample Requirements |
|
|
| Timeframe | Faster:
|
Slower:
|
| Technical Expertise | Moderate:
|
Advanced:
|
| Recommended Primary Methods |
|
|
| Recommended Secondary Methods |
|
|
- Evaluate Your Research Question:
Is your goal to develop a therapeutic antibody? → Prioritize conformational epitope mapping
Need to understand binding under denaturing conditions? → Choose linear epitope mapping
Developing diagnostics for detection of denatured proteins? → Linear epitope methods are sufficient
- Assess Available Resources:
Limited budget and equipment? → Start with peptide arrays or computational prediction
Access to specialized facilities and expertise? → Consider cryo-EM or X-ray crystallography
Moderate resources? → HDX-MS or XL-MS offer good balance of detail and accessibility
- Consider Time Constraints:
Need rapid results (days to weeks)? → Peptide arrays, phage display, or computational prediction
Can dedicate months to the project? → Structural approaches will provide highest resolution
Medium timeframe (weeks to months)? → HDX-MS, XL-MS, or alanine scanning
- Examine Antigen Properties:
Working with membrane proteins? → Cryo-EM is often most suitable
Small, soluble proteins? → X-ray crystallography may be ideal
Unknown structure? → Begin with computational prediction followed by experimental validation
- Optimal Integrated Approaches:
For linear epitopes: Computational prediction → peptide arrays → alanine scanning → functional validation
For conformational epitopes: Computational prediction → HDX-MS/XL-MS → mutagenesis → structural determination
At Creative Biostructure, we specialize in comprehensive epitope mapping services for both linear and conformational epitopes. With advanced platforms including cryo-EM and HDX-MS, our expert team delivers high-resolution insights to accelerate your antibody development, vaccine research, and diagnostic projects. Contact us to learn how our structural biology and protein engineering expertise can empower your discovery pipeline.
References
- Gershoni J M, Roitburd-Berman A, Siman-Tov D D, et al. Epitope mapping: the first step in developing epitope-based vaccines. BioDrugs, 2007, 21: 145-156. https://doi.org/10.2165/00063030-200721030-00002
- Nilvebrant J, Rockberg J. An introduction to epitope mapping. Epitope mapping protocols. New York, NY: Springer New York, 2018: 1-10.
- Francino-Urdaniz I M, Whitehead T A. An overview of methods for the structural and functional mapping of epitopes recognized by anti-SARS-CoV-2 antibodies. RSC Chemical Biology, 2021, 2(6): 1580-1589. https://doi.org/10.1039/D1CB00169H
- Grewal S, Hegde N, Yanow S K. Integrating machine learning to advance epitope mapping. Frontiers in Immunology, 2024, 15: 1463931. https://doi.org/10.3389/fimmu.2024.1463931