Combinational Applications of Cryo-EM and Mass Spectrometry
Determining the structure of large protein complexes at the molecular or atomic level and gaining insight into their function in the cellular environment has become a key part of modern structural biology. Conventional structural biology techniques, such as X-ray crystallography, NMR spectroscopy, and cryo-electron microscopy (cryo-EM), have their own advantages and applicable conditions, but when solving the structure of large dynamic complexes, it is necessary to integrate multiple complementary techniques. The combination of mass spectrometry with cryo-EM and integrative modeling is particularly promising for the study of the topology and structure of macromolecular complexes. The target structures solved by this integrated approach so far include ribosomes, proteasomes, chromatin remodelers, polymerases, and photosystem complexes.
Creative Biostructure combines cryo-EM with mass spectrometry, which can provide valuable structural information about large protein assemblies consist of components like proteins, post-translational protein modifications, nucleic acids, and lipids.
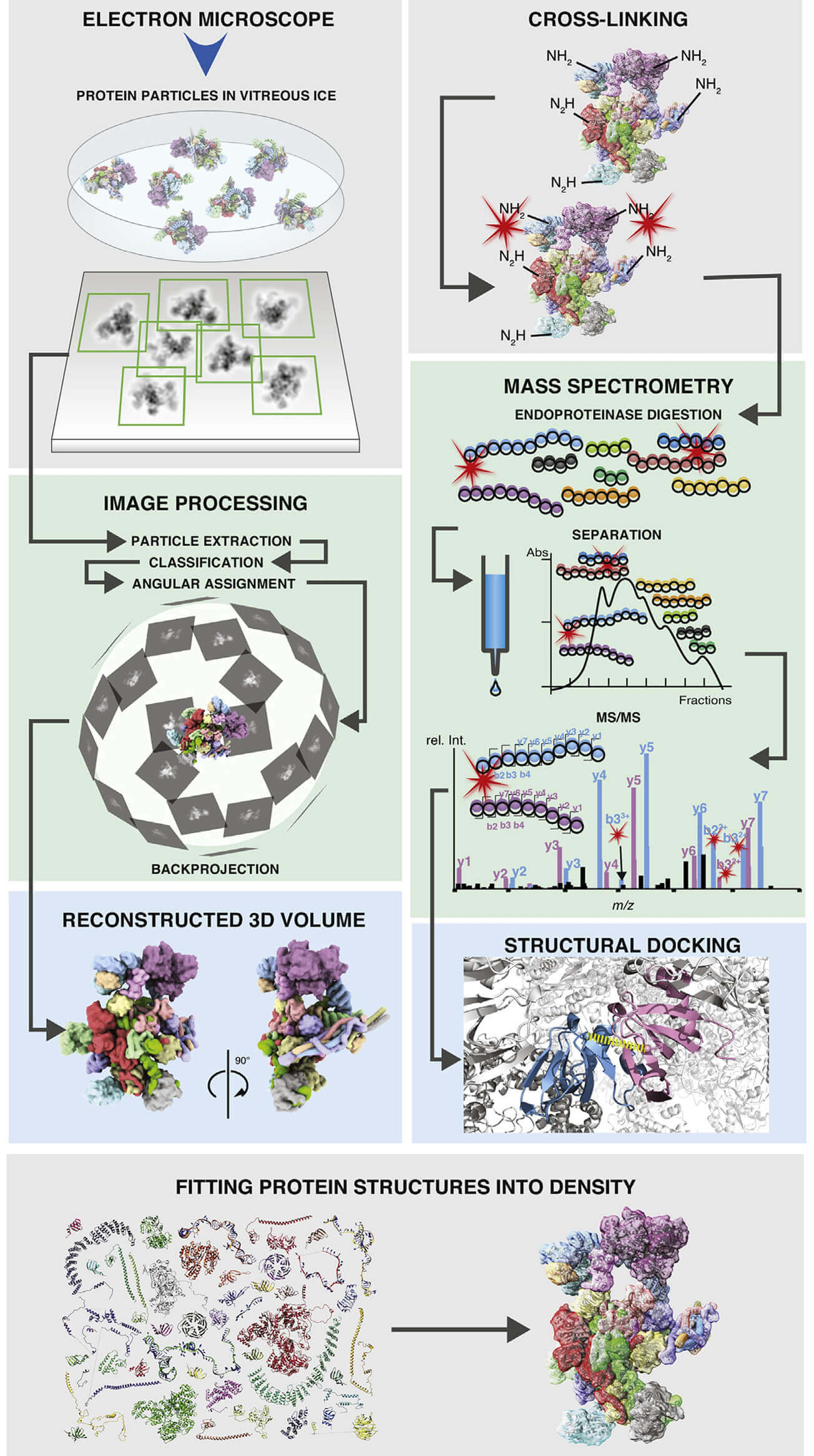 Figure 1. Schematic of workflows of single-particle cryo-EM and CX-MS of
macromolecular protein assemblies. (Schmidt C et al., 2017).
Figure 1. Schematic of workflows of single-particle cryo-EM and CX-MS of
macromolecular protein assemblies. (Schmidt C et al., 2017).
The Workflow of Combinational Applications of Cryo-EM and Mass Spectrometry
Following the basic process of integrative structural biology, our steps are as follows:
- Identification and gathering of structural components: mass spectrometry can offer the subunit composition of a complex and reveal subunit stoichiometry. With single particle analysis (SPA), electron microscopy density maps can be obtained. Interaction data between components are collected from mass spectrometry.
- Generation of a scoring function: available data from mass spectrometry techniques are translated into spatial restraints, which are combined into a scoring function. Multiple sampling of these functions yields a set of assembly models with varying degrees of consistency with the input data.
- Building a consensus 3D model: models are ranked by score and the optimal models are clustered by root mean square deviation (RMSD).
Mass spectrometry data can provide valuable information on the proximity between amino acid residues as distance constraints for de novo or homology modeling. To further improve the atomic model, mass spectrometry can locate flexible domains in macromolecular complexes, thereby facilitating the modeling of regions that are less well defined in cryo-EM images. Creative Biostructure can also integrate structural information from other different sources and methods to make the atomic model more reliable, more complete, and more accurate. For detailed information or a quotation, please feel free to contact us.
Ordering Process

References
- Leitner A, et al. Crosslinking and mass spectrometry: an integrated technology to understand the structure and function of molecular machines. Trends in Biochemical Sciences. 2016, 41(1): 20-32.
- Schmidt C, Urlaub H. Combining cryo-electron microscopy (cryo-EM) and cross-linking mass spectrometry (CX-MS) for structural elucidation of large protein assemblies. Current Opinion in Structural Biology. 2017, 46: 157-168.


