Comparison of X-ray Crystallography, NMR and EM
Structural biology integrates techniques from molecular biology, biochemistry, and biophysics to elucidate the molecular structures and dynamics of biologically significant molecules. Advances in instrumentation have significantly enhanced the field, enabling the analysis of complex biological molecules with remarkable ease and precision. Understanding the three-dimensional structures of proteins and protein complexes offers profound insights into the mechanisms of life and disease, facilitating the rational design of novel diagnostic and therapeutic agents. The primary techniques in structural biology are X-ray Crystallography, nuclear magnetic resonance (NMR), and cryo-electron microscopy (Cryo-EM). Each method has distinct advantages and limitations, making it essential to choose the appropriate technique based on the specific research objectives.
Based on the data provided by the RCSB Protein Data Bank (PDB) on the number of released structures per year, although the proportion has declined, X-ray crystallography remains the dominant technique in structural biology, accounting for the majority of structures released each year. NMR has made a much smaller contribution to the total number of structures, generally contributing less than 10% annually. The method is particularly valuable for the study of smaller proteins and complexes in solution. The use of EM, especially cryo-EM, has increased significantly in recent years. From being almost negligible in the early 2000s, its contribution has risen sharply, especially after 2015, to account for up to 40% of new structure deposits by 2023-2024.
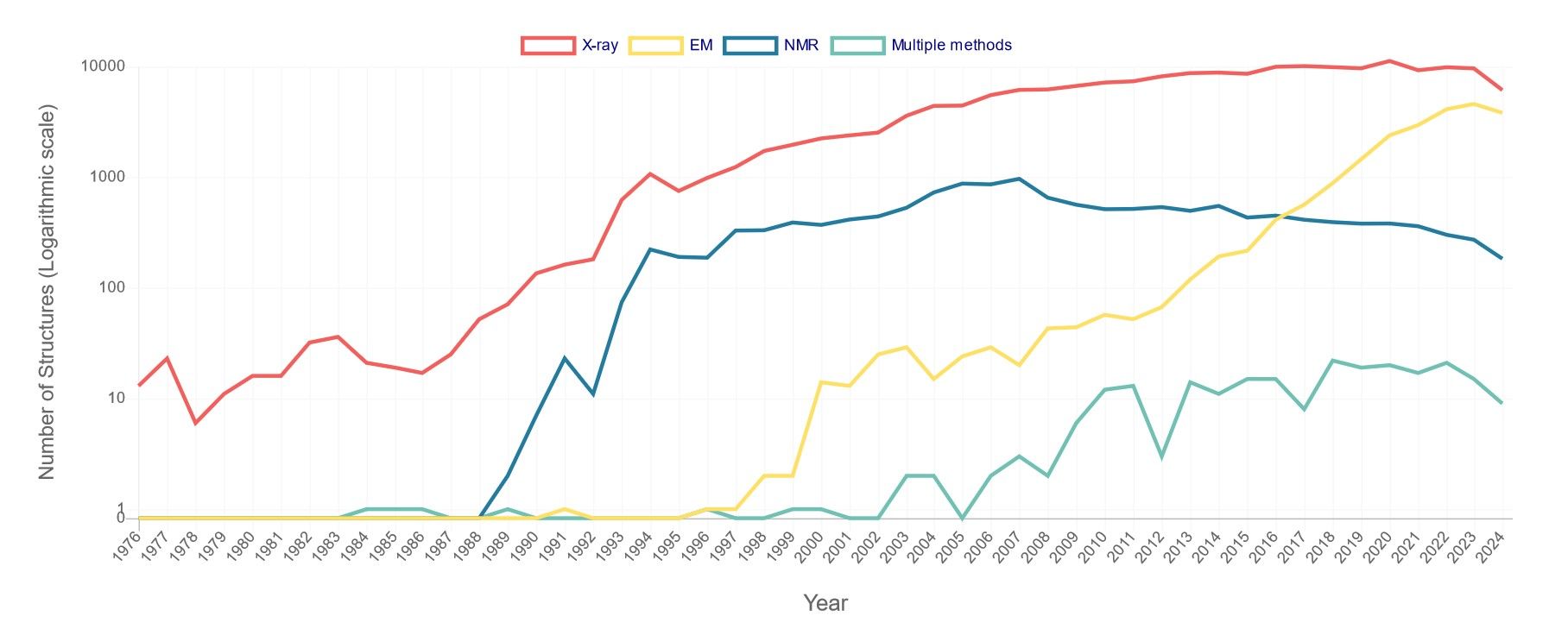 Figure 1. Number of PDB structures published per year. X-ray includes X-ray diffraction, fiber diffraction, or powder diffraction; NMR
refers to solution NMR or solid-state NMR; EM includes electron microscopy or electron crystallography or electron tomography; multiple methods means
multiple experimental methods. For example, if a structure is solved by X-ray diffraction and neutron diffraction, it is only counted in this category (Data
updated as of Sep 3rd, 2024).
Figure 1. Number of PDB structures published per year. X-ray includes X-ray diffraction, fiber diffraction, or powder diffraction; NMR
refers to solution NMR or solid-state NMR; EM includes electron microscopy or electron crystallography or electron tomography; multiple methods means
multiple experimental methods. For example, if a structure is solved by X-ray diffraction and neutron diffraction, it is only counted in this category (Data
updated as of Sep 3rd, 2024).
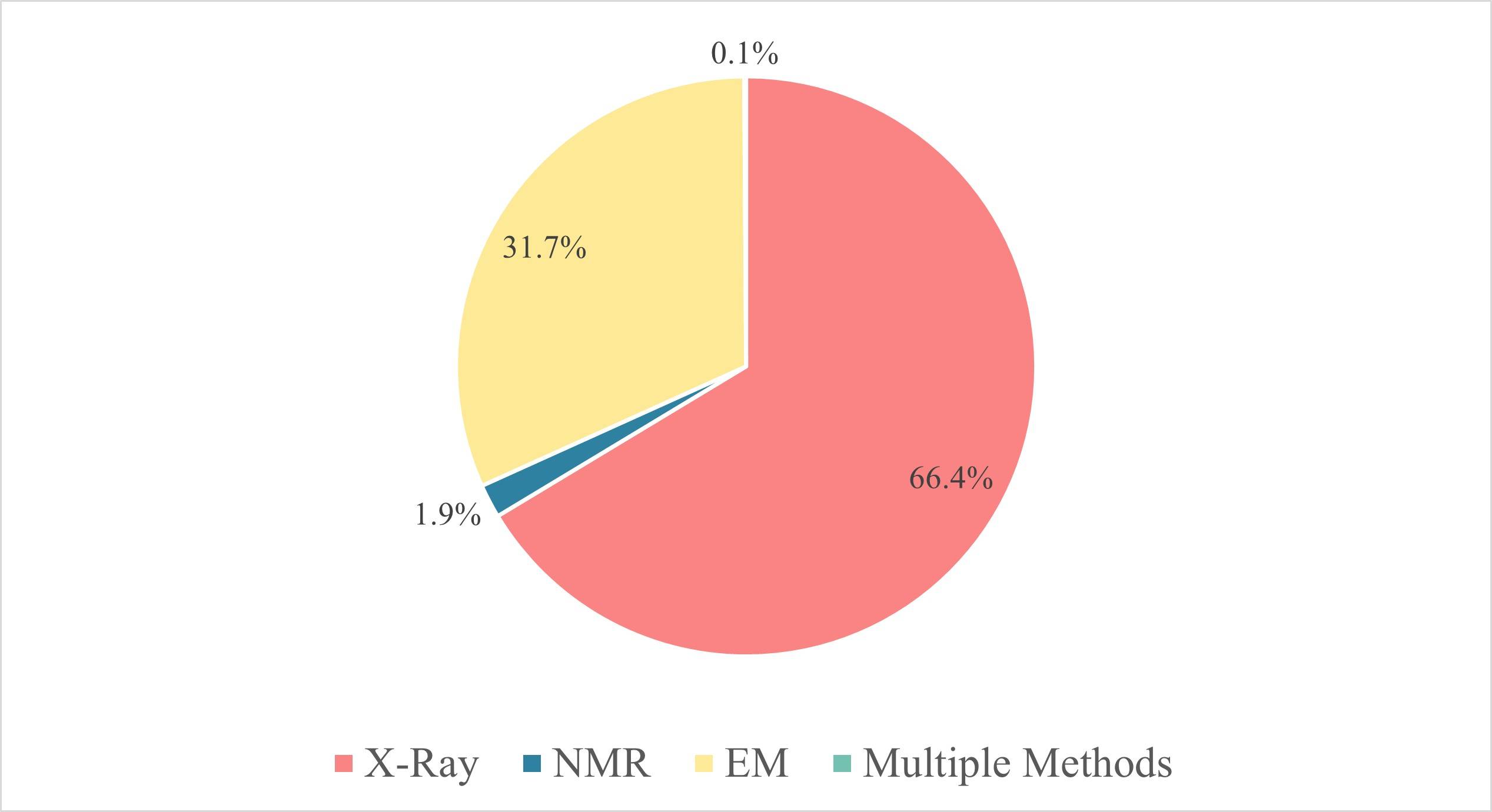 Figure 2. Three major research techniques in structural biology. According to the statistics of
PDB, in 2023, more than 9601 protein structures were solved by X-ray, accounting for more than 66% of the total. And there are 4579 protein structures
obtained by EM, accounting for 31.7%, and 272 structures obtained by NMR, accounting for only 1.9%.
Figure 2. Three major research techniques in structural biology. According to the statistics of
PDB, in 2023, more than 9601 protein structures were solved by X-ray, accounting for more than 66% of the total. And there are 4579 protein structures
obtained by EM, accounting for 31.7%, and 272 structures obtained by NMR, accounting for only 1.9%.
X-ray Crystallography
X-ray crystallography is a powerful and widely used technique in structural biology for determining the three-dimensional structures of biological macromolecules such as proteins, nucleic acids, and complex assemblies. By providing atomic-level resolution, X-ray crystallography has been instrumental in advancing our understanding of molecular mechanisms, functions, and interactions within cells. The technique is based on the diffraction of X-rays by the electron density of crystallized molecules, allowing researchers to construct detailed models of these structures. This method has been instrumental in numerous scientific discoveries, including the determination of the structure of DNA and the development of drugs that target specific proteins.
History and Actuality of X-ray Crystallography
X-ray crystallography originated in the early 20th century, following Wilhelm Conrad Röntgen's discovery of X-rays in 1895 and Max von Laue's demonstration of X-ray diffraction by crystals in 1912. Building on this, Sir William Henry Bragg and Sir William Lawrence Bragg developed X-ray crystallography as a method for determining crystal structure and formulated Bragg's Law to relate the angles of diffracted X-rays to the spacing between crystal planes. Their work led to the first atomic models of simple crystals, such as NaCl, and earned them the Nobel Prize in Physics in 1915.
The technique was later extended to biology, most notably leading to the determination of the structure of the DNA double helix by James Watson and Francis Crick in 1953, using X-ray diffraction data from Rosalind Franklin and Maurice Wilkins. Since then, X-ray crystallography has become an indispensable tool in structural biology, elucidating enzyme mechanisms, membrane protein structures, and large macromolecular complexes. Today, over 224,000 protein structures have been deposited in the Protein Data Bank, over 86% of which have been solved by X-ray crystallography, underscoring its dominance in the study of biological macromolecular structures.
Basic Theory of X-ray Crystallography
X-ray crystallography is based on the diffraction of X-rays by the electron clouds of atoms within a crystalline structure. When a crystal is exposed to a collimated beam of X-rays, the rays interact with the electrons in the crystal, leading to constructive and destructive interference. This interaction produces a diffraction pattern that can be recorded on a detector. The positions and intensities of the spots in the diffraction pattern are directly related to the electron density within the crystal.
Bragg's Law, nλ=2dsinϑ, where λ is the wavelength of the incident X-rays, d is the distance between crystal planes, ϑ is the angle of incidence, and n is an integer, describes the condition for constructive interference and is fundamental to the analysis of X-ray diffraction data. By measuring the angles and intensities of the diffracted beams, scientists can generate a three-dimensional map of the electron density within the crystal, which can then be interpreted to determine the positions of atoms.
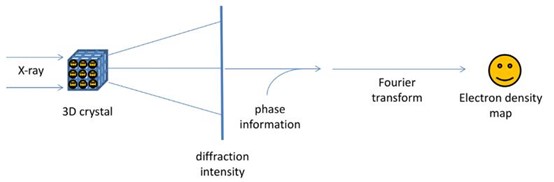 Figure 3. The physics and mathematical principles of X-ray crystallography to solve a structure.
Figure 3. The physics and mathematical principles of X-ray crystallography to solve a structure.
Process of X-ray Crystallography
The process of X-ray crystallography involves several key steps:
Crystallization: The target molecule, often a protein or nucleic acid, must be crystallized (protein crystallization). This step is often challenging, as obtaining high-quality crystals suitable for diffraction can require extensive screening and optimization of conditions.
Data Collection: The crystal is exposed to an X-ray beam, and the resulting diffraction pattern is recorded. Modern facilities often use synchrotron radiation sources, which provide intense and highly collimated X-rays, allowing for the collection of high-resolution data.
Data Processing: The diffraction data are processed to produce a set of structure factors that describe the amplitude and phase of each diffracted beam. Phasing is a critical step in this process because the phase information is not directly measurable and must be inferred using methods such as molecular replacement or experimental phasing techniques such as multi-wavelength anomalous dispersion (MAD) or single-wavelength anomalous dispersion (SAD) (phase determination).
Model Building and Refinement: An initial model of the molecule is built based on the electron density map generated from the processed data. This model is iteratively refined by adjusting atomic positions and validating the fit of the model to the experimental data, ultimately resulting in a detailed three-dimensional structure.
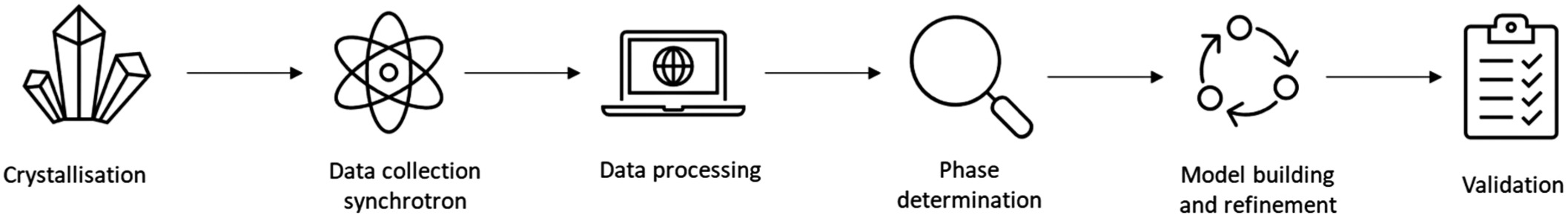 Figure 4. The process of X-ray crystallography (Bingham et al., 2023).
Figure 4. The process of X-ray crystallography (Bingham et al., 2023).
X-ray Crystallography vs. Single Crystal X-ray Diffraction (SC-XRD)
Single crystal X-ray diffraction (SC-XRD) and X-ray crystallography are closely related but are not exactly the same thing. In many circumstances, X-ray crystallography is often referred to as SC-XRD. More specifically, X-ray crystallography is the overarching technique, while SC-XRD is a specific application of this technique. SC-XRD is a subset of X-ray crystallography that focuses on single crystals, while X-ray crystallography is the general method that encompasses various techniques for studying crystal structures.
X-ray crystallography is a broad term that refers to the technique of determining the atomic structure of a crystal by measuring how X-rays are diffracted as they pass through the crystal. The resulting diffraction pattern provides information about the arrangement of atoms within the crystal, allowing scientists to reconstruct a detailed three-dimensional structure.
SC-XRD is a specific type of X-ray crystallography that focuses on single crystals, where the entire crystal is a single, continuous piece with a regular, repeating lattice structure. SC-XRD is the most commonly used form of X-ray crystallography for determining the detailed structure of small molecules, inorganic compounds, and macromolecules such as proteins.
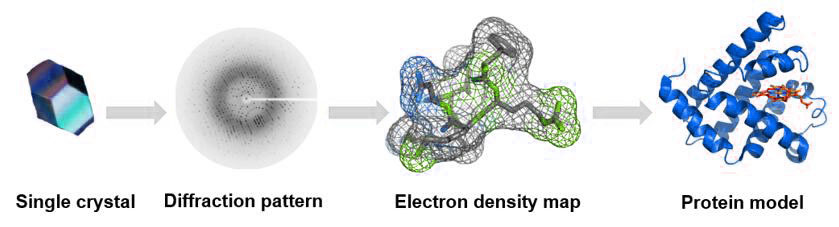 Figure 5: The process of single crystal X-ray diffraction technique.
Figure 5: The process of single crystal X-ray diffraction technique.
Different Types of Crystallography besides X-ray Crystallography
Crystallography does not always refer exclusively to X-ray crystallography. While X-ray crystallography is the most common and well-known form of crystallography, the term "crystallography" refers broadly to the study of crystal structures and can include a variety of techniques beyond X-ray methods. There are several types of crystallography.
X-ray crystallography is the most widely used technique for determining the atomic and molecular structure of crystals, particularly in structural biology, chemistry, and materials science. Neutron crystallography, using neutrons instead of X-rays, excels at locating light atoms like hydrogen, making it useful for studying hydrogen bonding and magnetic structures. Electron crystallography employs electron beams and is ideal for studying very small crystals, thin films, or two-dimensional materials, often in materials science and structural biology. Powder crystallography, a variant of X-ray crystallography, analyzes polycrystalline powders when single crystals are unavailable, aiding in phase identification. Time-resolved crystallography tracks structural changes in real-time, useful for studying dynamic processes like enzyme reactions or phase transitions.
Subdivision Techniques of X-ray Crystallography
X-ray crystallography has several subdivision techniques tailored to specific types of samples or experimental conditions. These techniques help to extend the applicability of X-ray crystallography to various types of crystals and molecular structures. Some of the key subdivision techniques include:
Powder X-ray Diffraction (PXRD)
Powder X-ray diffraction is used when single crystals are not available, and the sample consists of small, randomly oriented crystallites. Instead of a single crystal, a powder sample is exposed to X-rays, and the diffraction pattern is collected. PXRD is widely used in materials science, mineralogy, and for analyzing polymorphic forms of pharmaceuticals. It provides information about the crystal structure, phase identification, and crystallinity.
Small-Molecule X-ray Crystallography
This technique focuses on determining the structures of small organic and inorganic molecules. It typically requires high-quality single crystals, and the diffraction patterns provide very detailed information about molecular geometry. Small-molecule crystallography is used for precise structure determination in organic chemistry, drug discovery, and materials science.
Macromolecular X-ray Crystallography (MX)
This is the standard method for determining the structures of large biological molecules such as proteins, DNA, and RNA. Crystallization of macromolecules is more difficult, but it provides atomic-level details of biomolecular structures. MX is commonly used in structural biology to understand enzyme mechanisms, protein-ligand interactions, and drug targets.
Serial Femtosecond Crystallography (SFX)
SFX uses extremely short pulses of X-rays from X-ray free-electron lasers (XFELs) to collect diffraction data from crystals too small or too fragile for conventional X-ray crystallography. This allows data to be collected from nanocrystals or weakly diffracting samples without radiation damage. It is used to study dynamic processes, perform time-resolved studies, and obtain structures of challenging macromolecules that are difficult to crystallize in large forms.
Microcrystal Electron Diffraction (MicroED)
Although technically an electron microscopy technique, MicroED allows for X-ray-like diffraction data collection from tiny crystals using an electron beam. It is particularly useful for small macromolecular crystals that are too small for traditional X-ray methods. MicroED is used in structural biology to determine structures from microcrystals of proteins, peptides, and other small molecules (MicroED for structural analysis).
Anomalous Dispersion Methods (SAD/MAD)
Single-wavelength anomalous dispersion (SAD) and multiple-wavelength anomalous dispersion (MAD) techniques take advantage of the different scattering properties of atoms, especially metals such as selenium, to solve phase problems in X-ray crystallography. By tuning the X-ray wavelength to the absorption edge of specific atoms, these techniques allow easier structure determination. SAD and MAD are widely used in structural biology to solve the phase problem in macromolecular crystallography, especially for proteins and nucleic acids.
Advantages of X-ray Crystallography
X-ray crystallography offers several advantages:
High Resolution: It provides atomic-level resolution, making it possible to visualize the precise arrangement of atoms within a molecule.
Versatility: It can be applied to a wide range of biological molecules. It is suitable for water-soluble proteins, membrane proteins as well as macromolecular complexes.
Detailed Structural Information: It reveals detailed information about molecular interactions, conformational changes, and binding sites, which is crucial for understanding biological functions and designing targeted therapeutics.
Disadvantages of X-ray Crystallography
Despite its strengths, X-ray crystallography also has limitations:
Crystallization Requirement: The need for high-quality crystals can be a significant bottleneck, particularly for large, flexible, or membrane-associated molecules.
Static Picture: The technique typically provides a static snapshot of the molecule in a crystalline state, which may not fully represent its behavior in a dynamic, solution environment.
Phase Problem: The indirect measurement of phase information requires additional experimental or computational methods, which can complicate the structure determination process.
Applications of X-ray Crystallography
X-ray crystallography has a wide range of applications in structural biology and beyond:
Drug Design: It is a critical tool in structure-based drug design, allowing for the rational design of small molecules that can bind to specific protein targets.
Enzyme Mechanism Studies: By revealing the detailed architecture of active sites, X-ray crystallography provides insights into the catalytic mechanisms of enzymes.
Membrane Proteins Structures: Although challenging, the crystallization and structural determination of membrane proteins using X-ray crystallography have led to significant advances in our understanding of processes like signal transduction and ion transport.
Protein-Protein Interactions: The technique has been instrumental in characterizing large macromolecular complexes, such as the ribosome, elucidating how proteins interact with each other and with nucleic acids.
Nuclear magnetic resonance (NMR)
Nuclear magnetic resonance (NMR) spectroscopy is a powerful analytical technique used in structural biology to determine the three-dimensional structures of biomolecules in solution. NMR provides detailed information about the physical and chemical environment of atoms within a molecule, making it invaluable for studying the structure, dynamics, and interactions of proteins, nucleic acids, and other complex biomolecules. Unlike X-ray crystallography, which requires crystallization, NMR allows the study of biomolecules under conditions that closely mimic the physiological environment, providing insight into molecular behavior in the native state.
History and Actuality of NMR
Since the first observation of condensed state NMR signals in 1946, NMR technology has experienced rapid development for more than 70 years, and its application has been extended from the field of physics, such as nuclear magnetic moment determination, to chemistry, medicine, materials science, life science, and many others. In particular, in the 1980s, NMR technology was creatively applied to the structural analysis of proteins, thus promoting the application of NMR in the biological field. Although the amount of three-dimensional structural data of proteins obtained by NMR technology is not comparable to that of single crystal X-ray diffraction, the unique advantages of NMR technology have been widely recognized: NMR can provide information on a kinetic basis, so that the internal movement of proteins over multiple time scales and their binding mechanism to ligands can be resolved.
Basic Theory of NMR
NMR spectroscopy is based on the principle that certain atomic nuclei possess a magnetic moment due to their spin. When placed in a strong external magnetic field, these nuclei can absorb and re-emit electromagnetic radiation at specific frequencies, known as resonance frequencies. The resonance frequency is influenced by the magnetic environment of the nucleus, which is affected by the chemical structure and spatial arrangement of the atoms around it. By measuring these resonance frequencies, NMR provides information about the local environment of specific atoms within a molecule, allowing for the determination of molecular structure.
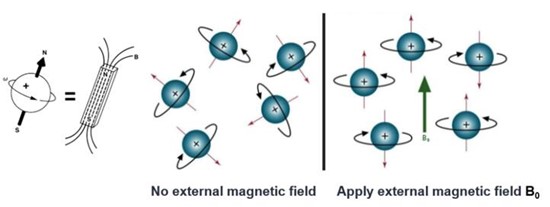 Figure 6. Nuclear spin without and with an external magnetic field.
Figure 6. Nuclear spin without and with an external magnetic field.
Process of NMR
The process of determining a molecular structure using NMR involves several key steps:
Sample Preparation: The biomolecule of interest is typically isotopically labeled with NMR-active nuclei such as 13C, 15N, or 2H to enhance signal sensitivity and resolution (NMR spectroscopy services).
Data Acquisition: The sample is placed in a strong magnetic field, and a series of radio frequency pulses are applied to excite the nuclei. The emitted signals are recorded as free induction decays (FIDs).
Spectral Processing: The FIDs are transformed into NMR spectra using Fourier transformation. The spectra provide information about the chemical environment of the nuclei. The signals in the NMR spectra are assigned to specific atoms in the molecule based on known chemical shifts and coupling patterns.
Structure Analysis: Using the assigned resonances and distance constraints derived from NOE (Nuclear Overhauser Effect) experiments, the three-dimensional structure of the molecule is calculated through computational algorithms.
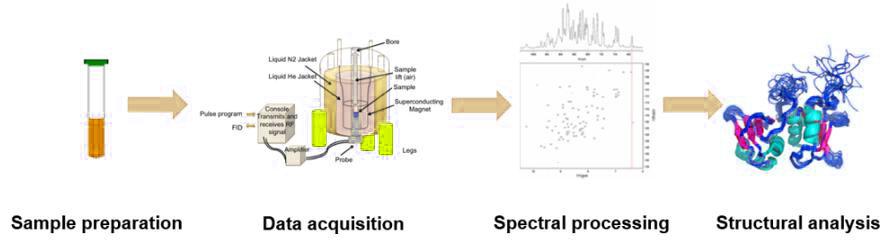 Figure 7. The process of nuclear magnetic resonance technology.
Figure 7. The process of nuclear magnetic resonance technology.
Subdivision Techniques of NMR
NMR spectroscopy includes several subdivision techniques, such as:
Multidimensional NMR: Multidimensional NMR techniques, such as 2D, 3D, and 4D NMR, spread spectral information across multiple dimensions, allowing for greater resolution of complex spectra. This approach helps overcome signal overlap problems that are common in large molecules such as proteins and nucleic acids. By correlating data from different dimensions, these experiments improve the ability to assign signals to individual atoms, providing a clearer picture of molecular structure. Multidimensional NMR is essential for the study of biomolecules where single-dimensional NMR lacks the resolution required for accurate structural interpretation.
Relaxation NMR: Relaxation NMR focuses on the analysis of relaxation times, T1 (spin-lattice relaxation) and T2 (spin-spin relaxation), to gain information about molecular motion and dynamics. These relaxation times reflect how quickly nuclear spins return to equilibrium after excitation, providing insight into molecular flexibility, internal motions, and interactions with the environment. Relaxation NMR is particularly valuable in the study of large biomolecules, providing information about their dynamic behavior and how this affects their biological activity.
NOE Spectroscopy: Nuclear Overhauser Effect (NOE) spectroscopy uses dipolar interactions between nearby atomic nuclei to measure distances between atoms. By observing changes in signal intensity due to these interactions, NOE provides critical distance constraints used to construct three-dimensional molecular structures. This technique is particularly important in structural biology, where it helps define the spatial arrangement of atoms in biomolecules such as proteins and DNA, enabling a deeper understanding of their function and interactions.
Advantages of NMR
Solution-State Analysis: NMR allows the study of biomolecules in solution, closely mimicking physiological conditions.
Dynamic Information: NMR provides insights into molecular dynamics, conformational flexibility, and interactions that are not accessible by static techniques like X-ray crystallography.
No Crystallization Required: NMR does not require crystallization, making it suitable for studying molecules that are difficult to crystallize.
Disadvantages of NMR
Size Limitation: NMR is limited by molecular size, with practical upper limits for structure determination around 50 kDa, although advancements are pushing these limits.
Sensitivity: NMR is less sensitive compared to other techniques, often requiring high sample concentrations and isotopic labeling.
Complexity: The interpretation of NMR data and structure determination can be complex and time-consuming, requiring specialized knowledge and software.
Applications of NMR
NMR spectroscopy is widely used in structural biology for:
Protein Structure Determination: NMR has been used to determine the structures of many proteins, including those that are membrane-bound or involved in signaling pathways.
Drug Discovery: NMR is used in fragment-based drug discovery, where it helps identify and optimize small molecules that bind to target proteins.
Biomolecular Interactions: NMR is used to study protein-ligand, protein-protein, and protein-nucleic acid interactions, providing insights into binding affinities and mechanisms.
Conformational Dynamics: NMR provides information on the dynamic behavior of biomolecules, revealing how conformational changes are related to biological function.
Electron Microscopy
Electron microscopy (EM) is a powerful imaging technique used in structural biology to visualize biological specimens at high resolution, often down to the atomic level. Unlike light microscopy, which is limited by the wavelength of visible light, EM uses electrons as the imaging source, allowing for much finer detail. EM has become indispensable for studying the structures of cells, viruses, proteins, and other biomolecular complexes. In particular, cryo-electron microscopy (cryo-EM), a technique in which samples are frozen in a near-native state, has revolutionized the field by allowing the visualization of biomolecules in their functional, hydrated forms.
History and Actuality of EM
The role of EM in structural biology has evolved dramatically over the decades. Initially, EM was used to study cellular structures and large viruses, but its application was limited by the damage caused by electron beams and the need for harsh sample preparation techniques. The introduction of cryo-EM in the 1970s alleviated these problems, allowing samples to be imaged in a near-native state without the need for chemical fixation or staining.
In recent years, cryo-EM has experienced a "resolution revolution" due to technological advancements, such as direct electron detectors and improved computational methods. These advancements have enabled the determination of high-resolution structures of macromolecular complexes, such as ribosomes, ion channels, and virus-like particles, with unprecedented detail. Cryo-EM has become a mainstream technique in structural biology, often used in conjunction with X-ray crystallography and NMR spectroscopy.
Basic Theory of EM
Electron microscopy relies on the interaction of a beam of electrons with a specimen to produce an image. Electrons have much shorter wavelengths than visible light, allowing EM to achieve much higher resolution. There are several types of electron microscopy, including TEM, Scanning Electron Microscopy (SEM), and Cryo-EM, each with its own specific applications and advantages:
Transmission Electron Microscopy (TEM): In TEM, a beam of electrons is transmitted through a thin specimen. The electrons that pass through the sample are collected to form an image, providing detailed information about the internal structure of the specimen.
Scanning Electron Microscopy (SEM): In SEM, a focused beam of electrons is scanned across the surface of a sample. The electrons scattered from the surface are detected to produce a high-resolution image of the topography of the sample.
Cryo-Electron Microscopy (Cryo-EM): Cryo-EM involves rapidly freezing the specimen in vitreous ice to preserve its native structure. Images are captured at different angles and combined to reconstruct a 3D model of the specimen.
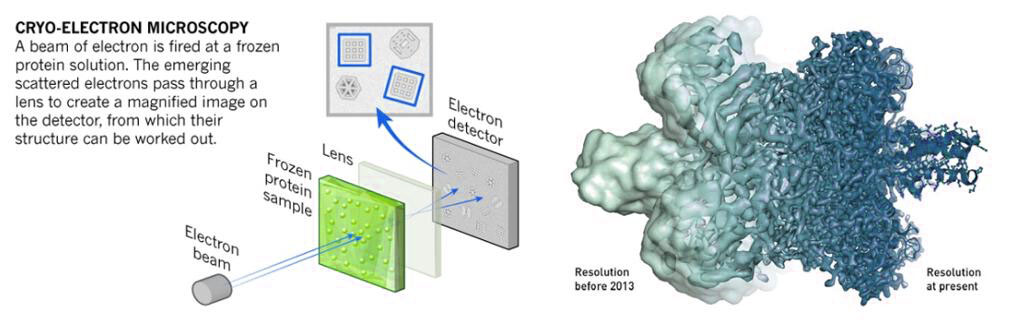 Figure 8. Mechanism of cryo-electron microscopy.
Figure 8. Mechanism of cryo-electron microscopy.
General Process of EM
The procedures involved in electron microscopy vary depending on the specific technique, but the general process includes the following steps:
Sample Preparation: The biological sample (e.g., proteins, viruses, or cells) is first suspended in a thin film of solution. A small amount of this sample is then applied to a specialized EM grid. Excess liquid is blotted away, leaving a thin layer of the sample. The grid is rapidly plunged into liquid ethane, freezing the sample in vitreous ice. This process preserves the sample in a near-native, hydrated state without the formation of ice crystals that could damage the structure.
Cryo-Transfer: The frozen grid is transferred to the cryo-electron microscope under cryogenic conditions, typically using a cryo-holder. This transfer ensures that the sample remains vitrified and stable at extremely low temperatures (around -180°C).
Data Collection: High-resolution images of the frozen sample are captured using an electron beam in the transmission electron microscope. Due to the low contrast of biological samples, direct electron detectors or other advanced cameras are used to capture images with enhanced sensitivity and resolution. Thousands of two-dimensional images of the sample are taken from different angles.
Image Processing: The collected images are processed using specialized software to correct for movement and reduce noise. Algorithms align the 2D images and classify them based on similar views of the particles. From these, thousands of particle images are selected and averaged to improve signal quality.
3D Reconstruction: Using computational techniques, the aligned 2D images are assembled into a three-dimensional model of the sample. This process involves reconstructing the structure by combining multiple particle views to create a high-resolution 3D map, which represents the electron density of the sample.
Model Building and Refinement: Based on the 3D map, a molecular model of the sample is built. The atomic structure is interpreted by fitting known atomic models (or ab initio models) into the density map. Further refinement is done to improve the accuracy of the structural model.
Validation and Analysis: Finally, the structure is validated using various metrics to ensure accuracy. The final cryo-EM structure is analyzed to understand the biological function, interactions, and mechanisms of the sample.
Subdivision Techniques of EM
Electron microscopy encompasses several subdivision techniques, including:
Single-Particle Analysis (SPA): Single-Particle Analysis (SPA) in cryo-electron microscopy (cryo-EM) is a method used to reconstruct the three-dimensional structure of biomolecules. It involves capturing thousands of 2D images of individual particles, such as proteins or viruses, in random orientations. These particles are then computationally aligned and averaged to produce a high-resolution 3D model. SPA is particularly useful for studying large, complex molecules, allowing researchers to visualize detailed structural features without the need for crystallization.
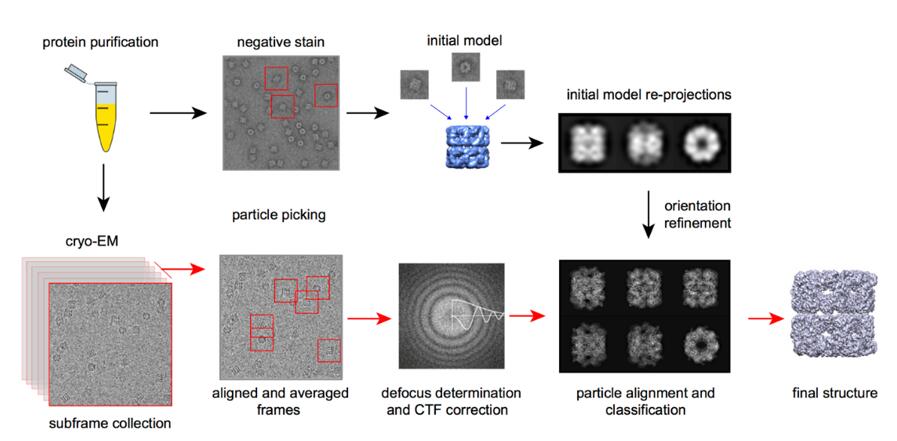 Figure 9. The process of Cryo-EM single particle analysis technique.
Figure 9. The process of Cryo-EM single particle analysis technique.
Electron Tomography: Electron tomography is a technique that provides three-dimensional reconstructions of cellular structures. By tilting the sample at various angles and capturing multiple images, a comprehensive set of projections is obtained. These projections are then combined computationally to create a 3D model of the specimen. Electron tomography is invaluable for studying the spatial organization of cellular components, providing detailed insight into subcellular structures and their relationships within the cell.
Cryo-Electron Tomography (Cryo-ET): Cryo-Electron Tomography (Cryo-ET) is an advanced version of electron tomography that integrates cryo-EM with tomography to study biological samples in their native, frozen-hydrated state. In Cryo-ET, a vitrified sample is tilted and imaged from multiple angles, allowing for the generation of 3D reconstructions of cellular structures in situ. This technique is especially powerful for visualizing the ultrastructure of cells and organelles, providing near-native, high-resolution insights into cellular architecture and molecular interactions.
Advantages of EM
High Resolution: EM, particularly cryo-EM, can achieve near-atomic resolution, allowing detailed visualization of macromolecular structures.
Versatility: EM can be used to study a wide range of biological specimens, from isolated proteins to entire cells.
Native State Imaging: Cryo-EM allows the visualization of biomolecules in their native, hydrated state without the need for staining or crystallization.
Disadvantages of EM
Radiation Damage: Biological specimens can be damaged by the electron beam, limiting the exposure time and the resolution that can be achieved.
Complex Sample Preparation: Sample preparation for EM, especially cryo-EM, can be technically challenging and time-consuming.
Cost and Accessibility: High-end electron microscopes and cryo-EM facilities are expensive and require specialized infrastructure and expertise.
Application of EM
Electron microscopy has a wide range of applications in structural biology:
Macromolecular Complexes: Cryo-EM is used to determine the structures of large protein complexes, such as ribosomes, polymerases, and membrane proteins.
Virus Structure: EM has been instrumental in revealing the structures of viruses, including the recent structure determinations of SARS-CoV-2 proteins.
Cellular Architecture: Electron tomography is used to visualize the architecture of cells and organelles in three dimensions, providing insights into cellular function and organization.
Drug Discovery: EM aids in drug discovery by revealing binding sites and conformational changes in target proteins, facilitating the design of new therapeutics.
Comparison of X-Ray Crystallography, NMR and Cryo-EM
In conclusion, each technology possesses distinctive advantages in specific applications. Consequently, one method may be employed extensively in certain instances, yet infrequently in others. Therefore, it is imperative to comprehend the nature of the analysis in order to select an appropriate method. Inappropriate method selection may not only yield compromised results but may also result in significant delays and financial losses. For further details, please refer to Table 1.
Table 1. The comparison of X-ray crystallography, NMR and Cryo-EM.
| Advantages | Disadvantages | Objects | Resolution | |
| X-ray Crystallography |
|
|
|
High |
| NMR |
|
|
|
High |
| Cryo-EM |
|
|
|
Relatively Low (<3.5 Å) |
Connections of X-Ray Crystallography, NMR and Cryo-EM
Integration of Techniques: These techniques are often used in combination to provide a more comprehensive understanding of molecular structures. For example, X-ray crystallography might be used to obtain a high-resolution structure of a protein, while NMR provides insights into its dynamics in solution, and cryo-EM reveals the structure of the protein within a larger complex.
Multi-Resolution Approach: Structural biologists often use a multi-resolution approach, combining data from cryo-EM, X-ray crystallography, and NMR to fill in gaps in understanding. For example, cryo-EM can provide an overall structure of a large complex, while X-ray crystallography or NMR are used to detail specific regions or dynamic interactions.
Structural Validation: The use of different methods allows cross-validation of structural data. A structure determined by cryo-EM can be compared with X-ray crystallography or NMR data to confirm its accuracy and to interpret functional aspects.
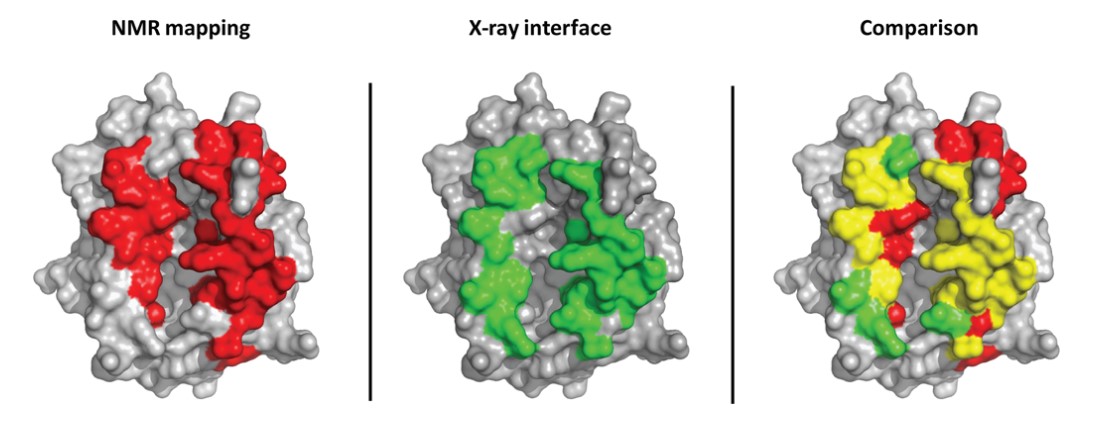 Figure 10. Comparison between an intermolecular interface determined by NMR mapping and X-ray crystallography (TCR/pMHC complex). The
MHC structure is shown in gray, surface representation. MHC residues, whose NMR signal is affected by TCR binding, are shown in red on the left; MHC
residues within 5A of the TCR in the X-ray structure are shown in green in the middle. Interface residues according to both NMR and Xray are shown in yellow
on the right. The rightmost structure shows good agreement between NMR and X-ray mapping. Some short-range allosteric effects can be seen in the top right,
red residues in the comparison picture (Bardelli et al., 2015).
Figure 10. Comparison between an intermolecular interface determined by NMR mapping and X-ray crystallography (TCR/pMHC complex). The
MHC structure is shown in gray, surface representation. MHC residues, whose NMR signal is affected by TCR binding, are shown in red on the left; MHC
residues within 5A of the TCR in the X-ray structure are shown in green in the middle. Interface residues according to both NMR and Xray are shown in yellow
on the right. The rightmost structure shows good agreement between NMR and X-ray mapping. Some short-range allosteric effects can be seen in the top right,
red residues in the comparison picture (Bardelli et al., 2015).
At Creative Biostructure, our team of highly skilled experts is dedicated to advancing your research with the most precise structural insights. With extensive experience and access to state-of-the-art technologies—including X-ray crystallography, NMR, and cryo-EM—we are uniquely positioned to select the optimal technique for your specific needs. Our commitment to accuracy and excellence ensures that you receive the most reliable structural data to propel your research forward. We invite you to contact with us for personalized, cutting-edge solutions tailored to your scientific goals.
References
- Bardelli, M., Livoti, E., Simonelli, L., Pedotti, M., Moraes, A., Valente, A. P., & Varani, L. (2015). Epitope mapping by solution NMR spectroscopy. Journal of Molecular Recognition, 28(6), 393–400.
- Bingham, M.; et al. Biophysical screening and characterisation in medicinal chemistry. Progress in Medicinal Chemistry (Vol. 62, pp. 61–104) 2023.
- Callaway, E. The revolution will not be crystallized: a new method sweeps through structural biology. Nature News 2015 525(7568): 172.
- Carroni M, Saibil R. Cryo electron microscopy to determine the structure of macromolecular complexes. Methods 2016, 95: 78-85.
- Rankin, N.; et al. The emergence of proton nuclear magnetic resonance metabolomics in the cardiovascular arena as viewed from a clinical perspective. Atherosclerosis 2014, 237(1): 287-300.
- Wang, H. W.; Wang, W. How cryo‐electron microscopy and X‐ray crystallography complement each other. Protein Science 2017, 26(1): 32-39.



